Fissaidee 3
| Sito: | Federica Web Learning - LMS |
| Corso: | Biologia di base |
| Unit: | Fissaidee 3 |
| Stampato da: | Utente ospite |
| Data: | martedì, 26 agosto 2025, 19:24 |
1. Autoassemblaggio delle proteine
Le informazioni necessarie per il ripiegamento del polipeptide sono presenti nella sua struttura primaria. Il polipeptide può quindi ripiegarsi spontaneamente per acquisire la conformazione proteica necessaria per l’attività biologica, come è stato dimostrato dagli esperimenti di Anfinsen sulla ribonucleasi pancreatica bovina.
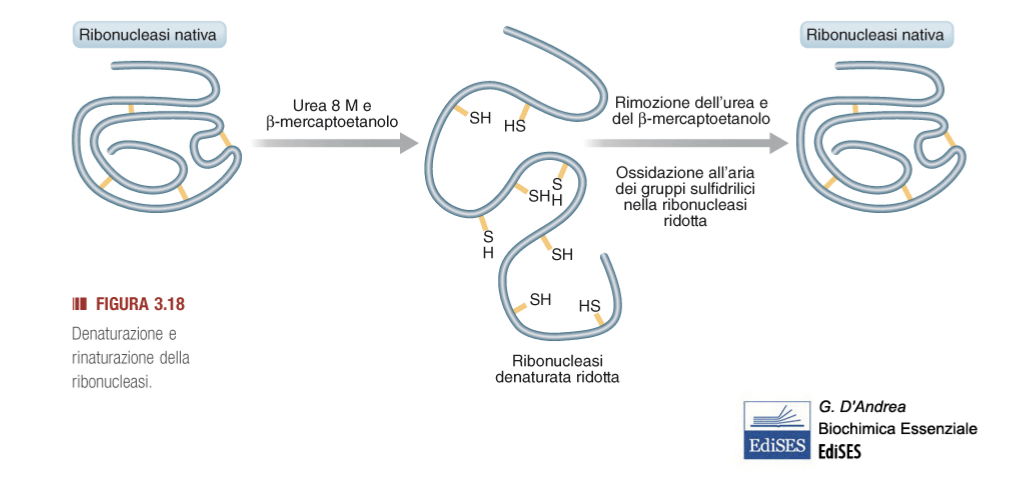
Il trattamento dell’enzima con agenti denaturanti e riducenti, che perturbano i legami tra i residui laterali degli amminoacidi, provoca la denaturazione della proteina con la perdita della sua attività funzionale, ma, in seguito all'allontanamento degli agenti denaturanti, la proteina è in grado di recuperare spontaneamente la sua conformazione nativa (rinaturazione). Pur essendo un processo spontaneo, l'autoassemblaggio avviene raramente; piuttosto nelle cellule agiscono dei fattori proteici, chiamati chaperon molecolari, che favoriscono il corretto ripiegamento delle proteine riducendo il numero di errori.
2. Chaperon molecolari
Sono proteine che svolgono un ruolo molto importante nel prevenire gli errori di ripiegamento che possono dar luogo a strutture prive di attività biologica. Le chaperonine sono una classe di chaperon molecolari formate da 14 subunità polipeptidiche disposte in due anelli sovrapposti che delimitano una cavità, nella quale viene accolto il polipeptide neosintetizzato durante il processo di ripiegamento. Ciò riduce la formazione di ripiegamenti scorretti.
3. Misfolding e Prioni
Le proteine non correttamente ripiegate sono normalmente eliminate dalla cellula, ma se questo non avviene, esse possono accumularsi sotto forma di strutture fibrillari insolubili (fibrille amiloidi) che si accumulano formando placche amiloidi, causa di gravi malattie neurodegenerative.

Ci sono almeno 20 differenti proteine non correlate tra loro che possono dar luogo alle fibrille amiloidi. Tra queste rientra la proteina prionica.
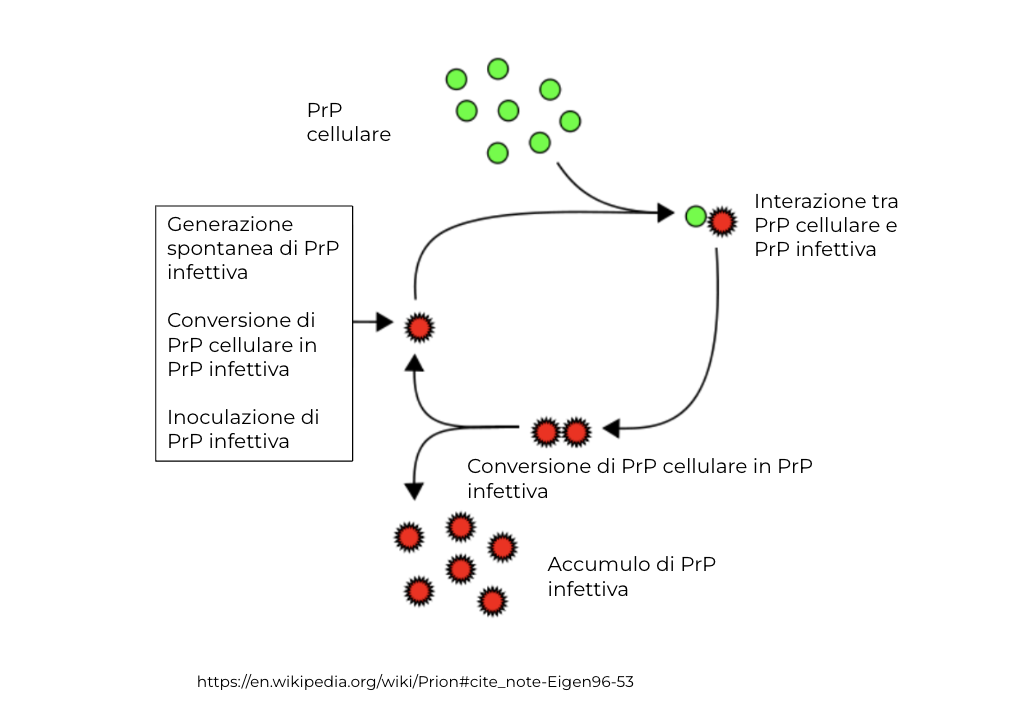
I prioni sono proteine che possono agire come agenti infettivi in alcune importanti malattie neurodegenerative note come malattie da prioni. Rappresentano un’eccezione al dogma universalmente accettato, in base al quale gli agenti infettivi devono possedere un proprio materiale genetico (DNA o RNA) in grado di propagare l’infezione nell’ospite.
Le malattie da prioni sono presenti negli animali e nell’uomo. Possono rimanere latenti per anni o per decenni, ma quando si manifestano sono spesso letali. La proteina prionica responsabile della malattia è una variante di una proteina prionica (PrP) normalmente presente nelle cellule del sistema nervoso centrale. La principale differenza tra la PrP normale e quella infettiva è di tipo conformazionale. Infatti, la PrP cellulare ha una struttura secondaria formata da diversi segmenti ripiegati ad alfa-elica che nella PrP infettiva si distendono assumendo la conformazione a foglietto beta.
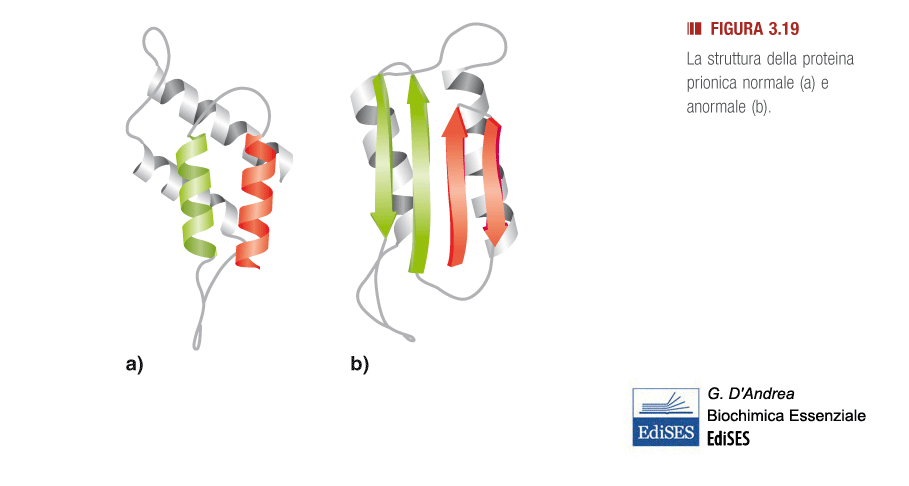
Le proteine PrP alterate si propagano rapidamente all’interno dei neuroni entrando in contatto con le molecole normali e provocandone la modificazione conformazionale che causa la malattia.