Fissaidee 3
3. Misfolding e Prioni
Le proteine non correttamente ripiegate sono normalmente eliminate dalla cellula, ma se questo non avviene, esse possono accumularsi sotto forma di strutture fibrillari insolubili (fibrille amiloidi) che si accumulano formando placche amiloidi, causa di gravi malattie neurodegenerative.

Ci sono almeno 20 differenti proteine non correlate tra loro che possono dar luogo alle fibrille amiloidi. Tra queste rientra la proteina prionica.
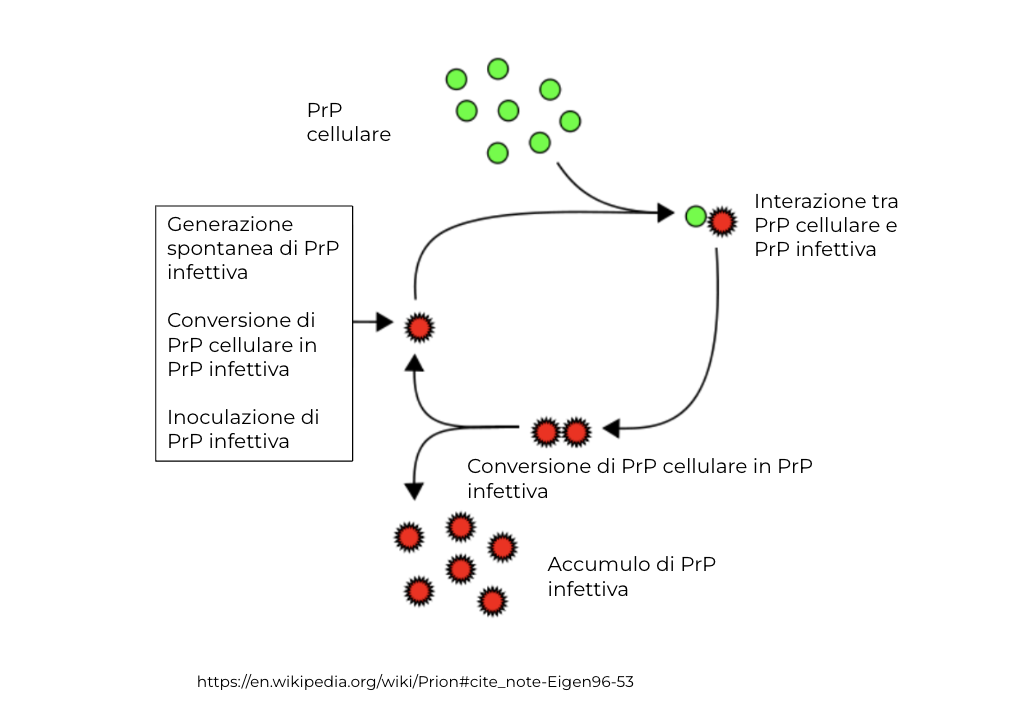
I prioni sono proteine che possono agire come agenti infettivi in alcune importanti malattie neurodegenerative note come malattie da prioni. Rappresentano un’eccezione al dogma universalmente accettato, in base al quale gli agenti infettivi devono possedere un proprio materiale genetico (DNA o RNA) in grado di propagare l’infezione nell’ospite.
Le malattie da prioni sono presenti negli animali e nell’uomo. Possono rimanere latenti per anni o per decenni, ma quando si manifestano sono spesso letali. La proteina prionica responsabile della malattia è una variante di una proteina prionica (PrP) normalmente presente nelle cellule del sistema nervoso centrale. La principale differenza tra la PrP normale e quella infettiva è di tipo conformazionale. Infatti, la PrP cellulare ha una struttura secondaria formata da diversi segmenti ripiegati ad alfa-elica che nella PrP infettiva si distendono assumendo la conformazione a foglietto beta.
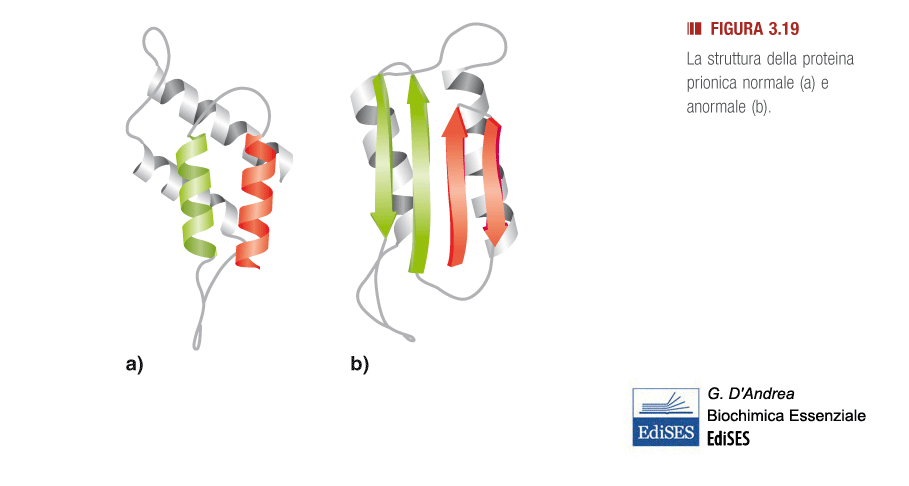
Le proteine PrP alterate si propagano rapidamente all’interno dei neuroni entrando in contatto con le molecole normali e provocandone la modificazione conformazionale che causa la malattia.